Thoughts on Structural Variation Detections on Metagenomes
We are now in the golden age of identifying genomic variation in population-scale studies. To date, much attention has focused on analyzing structural variation in primate genomes, specifically the human genome, given the direct relevance to human health. However, recent studies have shown that structural variations (SVs) are also common in microbial communities inhabiting human hosts and can affect host health. In fact, since the first bacterial genome Haemophilus Influenza was sequenced in 1995, genome-scale structural variations in bacterial chromosomes have been investigated, including inversions, duplications, rearrangements, and deletions. SVs in bacterial genomes have been repeatedly shown to not only have a major impact on function, but also linked to strong clinical relevance such as antimicrobial resistance, heteroresistance, and virulence. Given these findings, metagenomic studies have interrogated SVs within human host associated microbiomes.
Even very recently, structural variations in microbiomes have been linked to impacting human host health directly. In spite of these intriguing findings, SVs have not yet been comprehensively surveyed due to low resolution that short reads provide in the context of microbial communities. Also, the computational challenges in metagenomics like alignment and assembly errors require expert users to stay up to date with developments and best practice approaches. Although several methods have been proposed for SV detection in microbial genomes, these methods either are unable to detect all SV types or are limited with respect to the length of SVs they can detect. The arrival of the long read data era in metagenome sequencing provides enhanced resolution of SVs than previous short read data.
Here I want to list a table that include the major SV types that happens in bacteria and their machanisms, potential consequences and examples:
| SV type | Biological mechanisms | Potential consequences | Examples |
|---|---|---|---|
| INS | Horizontal gene transfer (HGT) | antimicrobial resistance | Extensive horizontal gene transfer in cheese-associated bacteria |
| Repeat expansion | virulence | ||
| Copy and paste | |||
| DUP | Tandem repeat: replication slippage and recombination | heteroresistance in Salmonella enterica | Tandem repeat (duplication): from several bps up to more than 200kbps |
| tRNA | methicillin resistance in Methicillin-Resistant Staphylococcus aureus | Nearly 300Kbp duplication of tRNA | |
| Segmental duplications: not relevant to the function of the duplicated sequence but common when homologous recombination happens | Amplification of β-lactamase gene can lead to de novo carbapenem resistance | E.coli DH10B: 113,260bp tandem duplication | |
| Segmental duplications: 1kbps upto 100-200 kbps but shorter than whole-genome duplication | |||
| DEL | Repeat contraction | Gene loss causes loss of functions or other functional consequences | several base pairs to several Mbps |
| Reductive genome evolution | R. rickettsii Iowa and R. rickettsii Sheila Smith | ||
| INV | homologous recombination, | antibiotic resistance gene | X-alignments between species |
| can be mediated by tyrosine site-specific recombinases | virulence gene | ||
| BND/TRA | Recombinations | change gene regulation | Depends on other SV events |
| By-product of other SV events | virulence | Mobile genetic elements: | |
| Transduction | Phage: transduction | ||
| Conjugation | Naked DNA: transformation | ||
| Transformation | Plasmid, chromosome: conjugation |
The current methods for detection SVs in metagenomics can be divided by sequencing technologies. Short read based methods usually use k-mer based or de bruijn graph-based approaches due to the limitation of the read lengths. On the other hand, long-read based methods can levearge the larger read length for better alignment or overlap graph building. Here I am attaching a figure that shows the high-level view of reference-based and assembled based methods for detection SVs in single-isolate bacterial genomes and metagenomes.
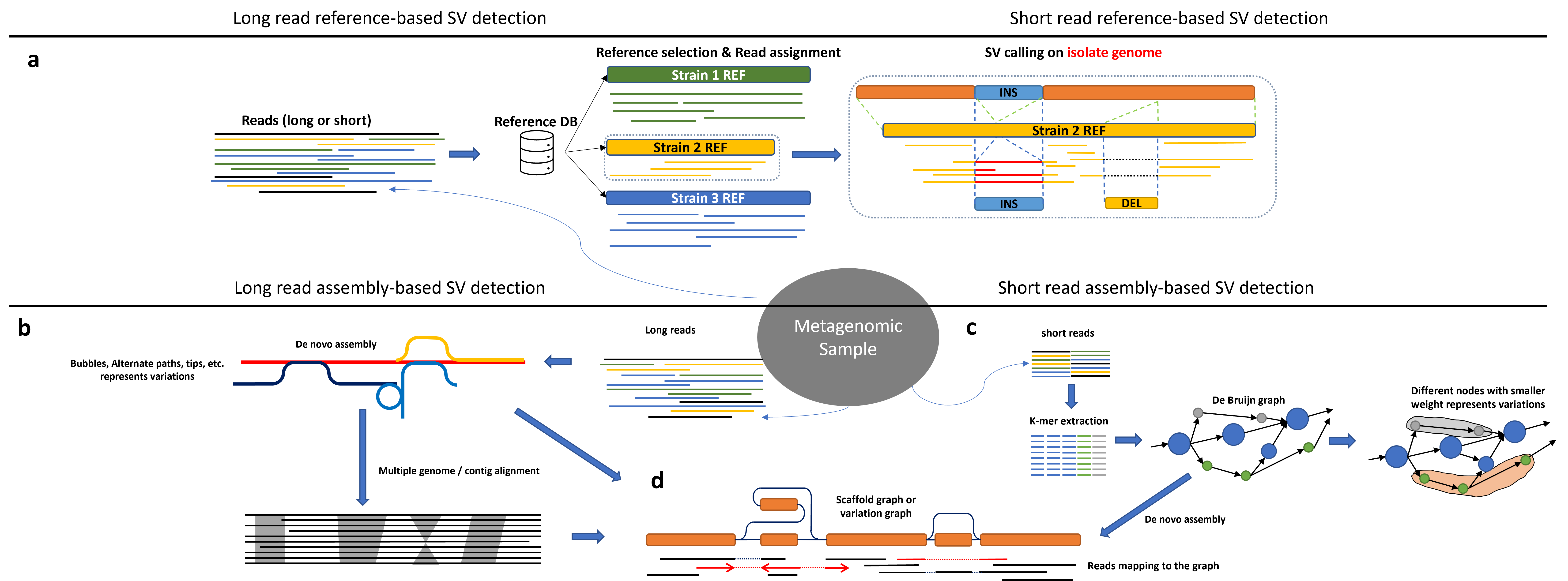 a: illustration of referenced-based SV detection methods among metagenomic sample and isolate genomes. For metagenomic samples given a read set, select proper reference genomes based on reads, then further perform split-read alignment on read set and references. The SV analysis step between reads and references is the same as SV calling on isolate genomes. b: Long read assembly-based methods can utilize multiple sequence alignment algorithms or graph-based algorithms onto contigs or assembly graphs. c: Short read de novo assembly usually includes k-mer counting. By generating de Bruijn graphs based on k-mers, graph-based analysis can indicate potential SV existence. d: A scaffold graph or variation graph can be inferred from either long read assembly graphs or short read assembly graphs.
a: illustration of referenced-based SV detection methods among metagenomic sample and isolate genomes. For metagenomic samples given a read set, select proper reference genomes based on reads, then further perform split-read alignment on read set and references. The SV analysis step between reads and references is the same as SV calling on isolate genomes. b: Long read assembly-based methods can utilize multiple sequence alignment algorithms or graph-based algorithms onto contigs or assembly graphs. c: Short read de novo assembly usually includes k-mer counting. By generating de Bruijn graphs based on k-mers, graph-based analysis can indicate potential SV existence. d: A scaffold graph or variation graph can be inferred from either long read assembly graphs or short read assembly graphs.
The methods includes these approaches are shown in the following chart.
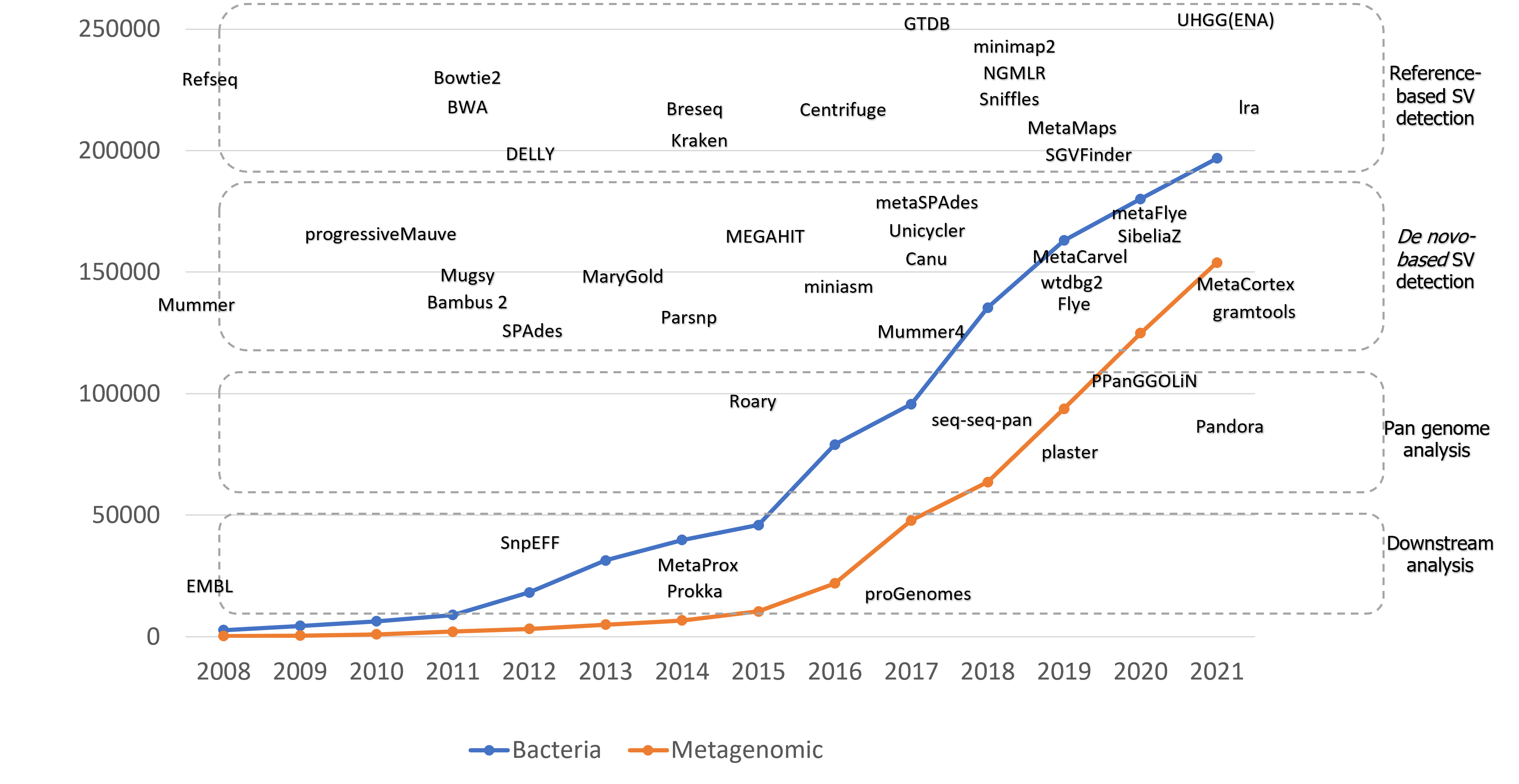
Till now, most of the SV detection methods in microbiomes are short-read based. However, the lack of long-range positional information brings the apparent need for long reads. Long reads can carry the ability to detect complex SVs and large size SV detection. For example, large tandem repeats extensively exist in e.coli, and those repeats can significantly improve the detection of those with the introduction of long reads. Validation is also a vital step of method detection, especially the methods that aim to solve microbiomes’ problems, given the uncertainty. Similar to the metagenome classification problem, a benchmarking dataset provides significant help for scientists to validate their methods. However, such datasets do not exist in SV detection in microbiomes. Thus the effectiveness of methods is hard to determine.